Nature Metabolism:NK细胞—脂肪组织代谢轴驱动抗病毒免疫新机制
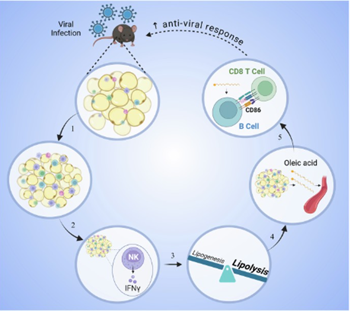
NK细胞来源的IFNγ动员脂肪组织中游离脂肪酸以促进病毒感染早期的B细胞激活
2025年4月,克罗地亚里耶卡大学等单位的研究人员在《Nature Metabolism》(IF:18.9)上发表了题为“NK cell-derived IFNγ mobilizes free fatty acids from adipose tissue to promote early B cell activation during viral infection”的研究论文,该研究揭示了一种先前未被重视的感染代谢适应机制,为病毒感染期间及脂肪组织炎症背景下的免疫-脂肪相互作用机制提供了新的见解。

-
病毒感染诱导脂肪组织上调NKG2D配体,促进自然杀伤(NK)细胞产生干扰素γ(IFNγ)。 -
IFNγ直接刺激脂肪细胞,使其从脂质合成转向脂解作用,导致循环中脂质释放,特别是游离脂肪酸(FFAs)。 -
油酸(OA)促进了B细胞上共刺激分子B7的表达,并增强了其激活CD8+ T细胞的能力。
研究背景:
病毒感染会引发一系列全身性代谢适应,如发热、恶心、疲劳和食欲不振。尽管我们通常将这些变化视为病理现象,但它们实际上是由激活的免疫系统介导的一系列精细调控的改变,被称为疾病代谢。感染期间免疫系统会大幅增加对营养物质的消耗,以满足其代谢需求。然而,为何病毒感染会促使免疫系统抑制营养摄入并优先动用脂质储备(这种代谢状态通常与禁食相关),仍是一个尚未解决的悖论。
脂肪组织曾长期被视为惰性的脂质储存器官,但如今已被确认为调节全身稳态的关键角色。其代谢受到包括由免疫系统产生的各种因素的严格调控。脂肪组织中存在大量免疫细胞,这些细胞通过分泌抗炎细胞因子来维持稳态。在炎症条件下,脂肪组织驻留免疫系统的状态会完全改变,这在肥胖背景下得到了最充分的研究,即肥胖诱导的脂肪细胞应激会导致配体的表达,这些配体结合并激活NK细胞上的受体。因此,NK细胞产生细胞因子IFNγ,将巨噬细胞极化为M1样状态。这导致它们产生促炎细胞因子,进一步促进炎症并导致全身胰岛素抵抗。虽然肥胖诱导的脂肪组织炎症显然是一种病理反应,但其潜在机制与人们对病毒感染的反应有很大的相似之处。然而,目前尚不清楚这种反应在哪些条件下是有益的。
已有研究表明,严重病毒感染可诱发恶病质并伴随脂肪组织损失。于是研究人员进一步探究了轻度病毒感染是否也会对脂肪组织的营养成分产生影响。利用小鼠巨细胞病毒(mCMV)感染小鼠,并通过组织学方法分析脂肪组织随时间的变化。结果发现雄性和雌性小鼠的平均脂肪细胞的大小显著减少,该现象在感染后3天最为显著。不过这种效应是短暂的,因为脂肪细胞在感染后7天即恢复原有大小且脂肪减少是内脏脂肪组织(VAT)所特有的。因此,轻度感染会导致脂肪细胞营养成分的暂时性丧失。
为探究脂肪组织的减少是否伴随脂肪衍生代谢物在循环系统中的增加,研究人员采用质谱技术分析了血浆脂质组成。结果显示,感染动物的血浆中多种脂质种类显著增加,特别是FFAs、磷脂酰胆碱和磷脂酰乙醇胺。另外,主要储存于脂肪组织的胆固醇在感染动物血清中也出现升高。这些数据表明,mCMV感染可能促使营养物质从脂肪组织向血液转移。使用RNA病毒(淋巴细胞性脉络丛脑膜炎病毒和甲型流感病毒)感染的小鼠进一步研究证实,脂肪细胞营养外流是对轻度和重度病毒感染的普遍反应。此外,通过RNA测序等结果表明,病毒感染初期会诱发一种疾病代谢状态,促使脂肪细胞代谢转向脂肪分解模式,从而导致营养物质从脂肪组织中流出且这一过程不依赖于全身营养物质的可用性。

病毒感染导致内脏脂肪细胞体积变小
鉴于病毒感染会诱导脂肪组织发生显著的形态学改变,研究人员推测这是感染过程中产生的特定促炎细胞因子的结果。转录组分析显示脂肪细胞中IFNγ应答基因表达上调;mCMV感染模型实验证实了中和IFNγ几乎完全阻断感染诱导的循环脂质增加,表明IFNγ直接介导了脂肪细胞的脂质外排。此外,通过构建AdiqCreIfngrFL/FL(IfngrΔAdi)基因敲除小鼠并在感染后第3天对纯化的脂肪细胞进行转录组分析,以探讨IFNγ是否直接靶向脂肪细胞并在感染后调节脂质代谢。结果显示与IfngrFL/FL同窝对照相比,感染IfngrΔAdi小鼠脂肪组织中IFNγ信号相关炎症分子表达降低。此外,IFNγ受体缺失阻断了脂肪细胞向脂解的代谢转变,具体表现为葡萄糖和FFA转运体表达升高以及参与甘油三酯分解代谢的基因表达降低。值得注意的是,血清脂质组分析显示,感染的IfngrΔAdi小鼠的血清脂质水平,特别是FFAs,显著低于IfngrFL/FL同窝对照。综上所述,感染诱导产生的IFNγ直接靶向脂肪细胞,介导其从脂质合成向脂解代谢的转变,从而导致脂肪细胞特性减弱及脂质含量的净流失。随后,研究人员通过IFNγ缺陷小鼠实验和流式细胞术等分析还证明了NK细胞通过NKG2D受体识别脂肪组织应激信号并随后分泌IFNγ,从而介导感染后脂肪细胞体积的减小。
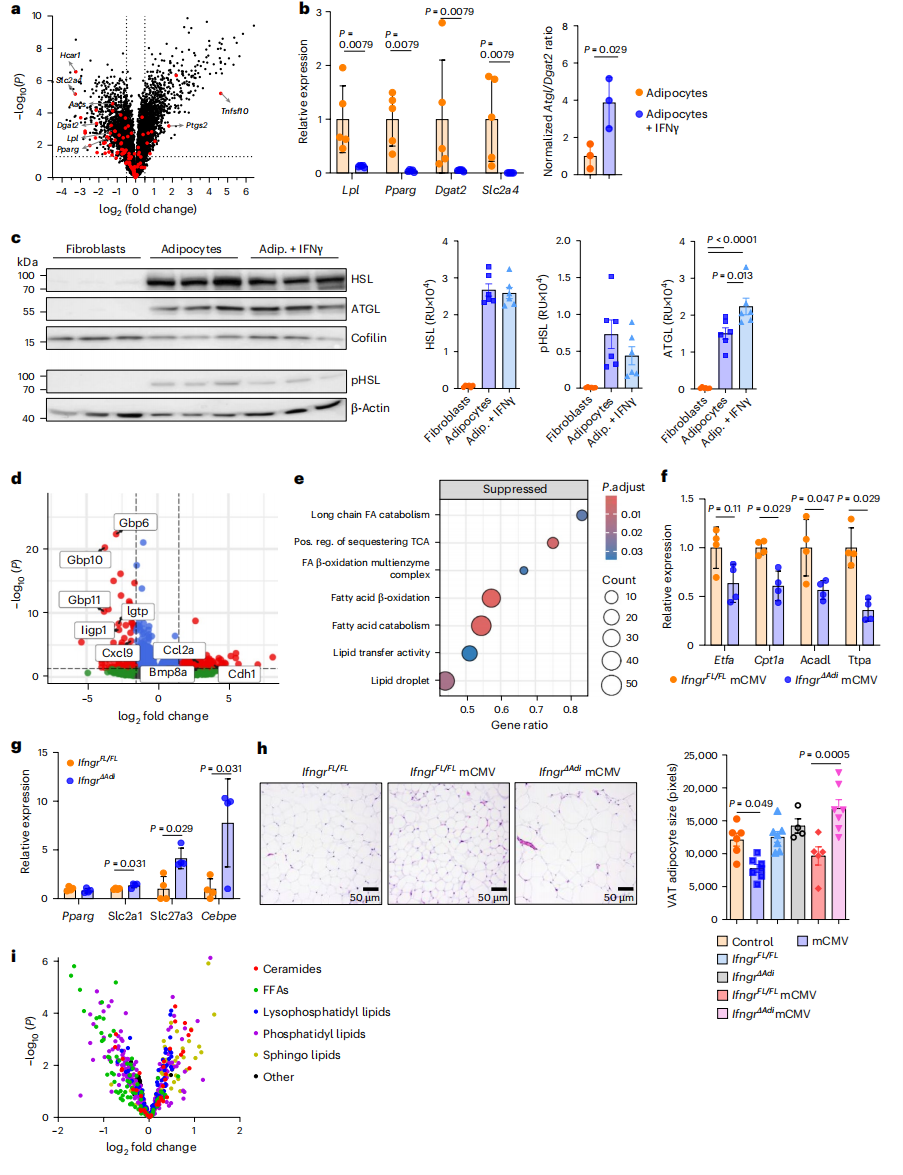
IFNγ直接靶向脂肪细胞并促使其代谢向脂解方向转变
接下来,研究人员探讨了感染诱导脂肪组织脂质释放的生理益处。血浆脂质组结果分析显示,感染动物血浆中FFAs,特别是油酸(OA)水平显著升高,而IFNγ中和处理可阻断该现象。随后,测试了几种循环中发现的最丰富的脂肪酸对A20 B淋巴瘤细胞共刺激B7分子表达的影响,其中,只有OA引起A20细胞表面CD80和CD86表达的增加。机制研究表明,OA能特异性增强基础呼吸、ATP合成和最大呼吸能力,阻断呼吸电子传递链或阻止线粒体外膜转运均可阻止OA介导的CD86对B细胞的诱导作用。因此得出结论:OA促进活化B细胞的呼吸代谢,这有利于共刺激分子的诱导,且这一现象在人类B细胞中也得了到验证。此外,与经OA预处理的A20 B细胞共培养的OT-1 T细胞(OT-1 T淋巴细胞是表达对该抗原特异性的转基因T细胞受体的CD8+ T细胞)表现出更高的CD69和CD25表达,并增强了产生TNF、IFNγ和颗粒酶B的能力,这种作用取决于CD86。总之,这部分结果说明了OA通过CD86激活B细胞,促进体外CD8+ T细胞启动。

脂肪酸通过调节氧化代谢促进B细胞体外共刺激潜能
研究结果表明,病毒感染后早期激活B细胞共刺激能力的增强源于循环系统中脂质利用度的提高。为验证这一机制,研究人员使用一种小分子抑制剂阿昔莫司进行体内实验,它可以阻止脂肪组织中的脂肪分解并降低血脂。结果观察到在感染3天后,阿昔莫司阻止了雄性小鼠早期激活的B细胞上CD86的诱导并显著削弱了CD8+ T细胞反应,导致感染后后期病毒滴度增加。最后,混合骨髓嵌合体实验表明,早期激活的B细胞中FFA摄取受损会损害其共刺激CD8+ T细胞的能力。总之,病毒感染引起VAT中脂肪细胞代谢的变化,这是由NK细胞产生的IFNγ介导的,导致脂质释放到循环中。循环中FFA可用性的增加促进了早期激活的B细胞的共刺激能力,从而增强了抗病毒CD8+ T细胞反应。

感染诱导的脂质释放促进体内B细胞介导的T细胞活化
总之,这项研究确定了通过NK细胞产生的IFNγ动员脂肪组织中的FFA,促进B细胞的早期激活,并增强CD8+ T细胞的抗病毒反应。这些发现为理解免疫细胞与脂肪组织在炎症反应中的相互作用提供了新的视角,并可能对开发新的抗病毒治疗策略具有重要意义。