


Nature Metabolism:癌症恶病质治疗曙光:PDK3能否成为关键“救星”?
肝糖异生和PDK3上调驱动果蝇和小鼠的癌症恶病质
2025年4月,哈佛医学院等单位的相关研究人员在《Nature Metabolism》(IF:18.9)上发表了题为“Hepatic gluconeogenesis and PDK3 upregulation drive cancer cachexia in flies and mice”的研究论文,发现了一个以前未知的与肝脏丙酮酸脱氢酶激酶同工酶3(PDK3)表达有关的致病机制,表明肝脏中的PDK3可能是癌症恶病质的潜在治疗靶点。

亮点概述:
- 脂肪体糖异生增加Yki果蝇的海藻糖水平。
- JAK-STAT通路刺激脂肪体糖异生。
- 抑制脂肪体中的JAK-STAT可缓解恶病质症状。
- IL-6-JAK-STAT信号介导Pck1和Pdk3在肝脏中表达,这些基因在果蝇、小鼠和人类中的表达增加与预后不良有关。
研究背景:
超过40%的癌症患者会出现恶病质,这是一种由肿瘤导致的危及生命的病症,其症状包括体重大幅下降、全身发炎、虚弱和疲劳。这些恶病质症状的一个主要驱动因素是肿瘤引起的代谢失调,如葡萄糖代谢的系统重编程。葡萄糖生成需要多种酶,如磷酸烯醇丙酮酸羧激酶(PEPCK)、丙酮酸羧化酶、果糖1,6-二磷酸酶(FBP)和葡萄糖6-磷酸酶(G6P),通过一系列反应从草酰乙酸合成葡萄糖。在这些酶中,PEPCK是限速酶,催化葡萄糖生成的第一步,将草酰乙酸转化为磷酸烯醇丙酮酸,以生成葡萄糖。除了直接参与糖异生的酶外,丙酮酸脱氢酶激酶(PDK)可能在促进糖异生方面发挥关键作用。当糖异生被激活时,PDK通过使丙酮酸脱氢酶复合物(PDC)失活来抑制丙酮酸向乙酰辅酶A的转化,从而使丙酮酸转向丙酮酸羧化酶以产生草酰乙酸和糖异生。值得注意的是,无论是在正常、饥饿还是糖尿病情况下,人类和啮齿动物的肝脏中几乎检测不到Pdk3的表达,这表明Pdk3具有独特的调节机制和生理作用。
近年来,果蝇中出现了几种器官消耗和恶病质模型。特别是,Yorkie癌基因(Yki,又称Yap)在成年肠干细胞(ISCs)中的激活形式表达会产生与恶病质特性相关的肿瘤。这些肿瘤至少分泌四种因子:蜕皮激素诱导基因L2(ImpL2)、PDGF和VEGF相关因子1(Pvf1)、离子转运肽(ITP)和未配对3(Upd3)。白细胞介素-6(IL-6)是一种特别相关的炎症细胞因子,它能激活JAK-STAT信号通路,已被证明与癌症患者的体重减轻和死亡率呈正相关。Upd3是IL-6的果蝇同源物,可诱导外周组织中ImpL2的表达,从而影响胰岛素信号传导并导致身体消耗。
在哺乳动物中,G6P催化糖异生的最后一步产生葡萄糖。然而,在果蝇中,它的作用是由海藻糖-6-磷酸合成酶1(Tps1)完成的,它产生海藻糖而不是葡萄糖。为了研究脂肪体Pepck1和Pdk在调节全身碳水化合物水平中的作用,研究人员使用GAL4-UAS和LexA-LexAop双元系统选择性地降低它们在Yki果蝇脂肪体中的表达,分别操纵肠道和脂肪体中的基因表达。使用LexAop-Yki系,发现Yki果蝇中全身海藻糖水平显著降低,而葡萄糖和其他分析代谢物的水平不受Pepck1或Pdk缺失的影响。为了进一步证实糖异生在Yki果蝇中的作用,通过追踪13C3标记的丙氨酸,这是最具糖异生特性的氨基酸。正如预期的那样,在Yki果蝇中检测到13C3标记的草酰乙酸和磷酸烯醇丙酮酸的比例增加,而这些比例被脂肪体Pdk消耗显著消除。总之,这些发现表明,由Pepck1和Pdk上调诱导的脂肪体糖异生导致Yki果蝇海藻糖水平升高。

Yki果蝇脂肪体中Pepck1和Pdk的上调刺激糖异生
接下来,研究人员探究了Upd3-JAK-STAT通路的作用。Yki果蝇的全身转录组分析显示,脂肪体是JAK-STAT信号显著激活的组织之一,其靶基因Socs36E的上调证明这一点。与此一致的是,在野生型果蝇ISCs中过表达Upd3会增加脂肪体Pepck1和Pdk的表达。值得注意的是,两个独立的染色质免疫沉淀高通量测序(ChIP-seq)数据集分析显示Pepck1和Pdk中存在JAK-STAT通路转录因子Stat92e的潜在结合位点。为了直接评估JAK-STAT通路对Pepck1和Pdk的调控,研究人员在脂肪体中表达了一种标记的、组成型活性的Stat92e。与这些区域存在多个STAT结合基元一致,ChIP显示Stat92e在脂肪体中与Pepck1和Pdk存在物理结合。这些观察结果表明,JAK-STAT信号直接促进脂肪体Pepck1和Pdk的表达,这是在Yki果蝇中观察到的糖异生增加所必需的。
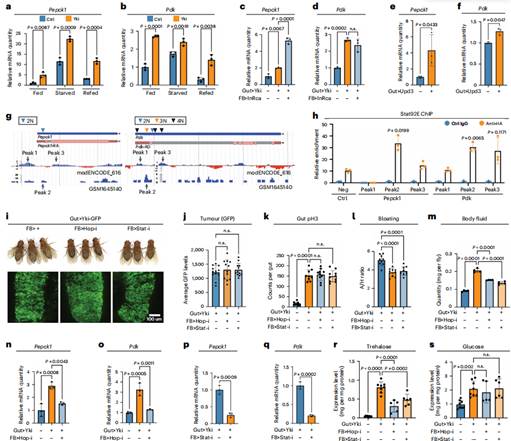
JAK-STAT信号通路调节脂肪体中Pepck1和Pdk的表达
鉴于癌症恶病质是影响健康和生存的主要因素,研究人员试图确定抑制Yki果蝇脂肪体JAK-STAT信号是否可以抑制恶病质症状。结果发现抑制Yki果蝇脂肪体中的JAK-STAT信号(通过hop/JAK缺失)和糖异生(通过Pdk缺失)都能恢复果蝇爬升能力并提高总存活率。
糖异生由非碳水化合物碳底物(如乳酸和糖原氨基酸)产生海藻糖和葡萄糖,研究人员假设糖异生的增加导致这些底物水平的不平衡。为了验证这一点,研究人员使用代谢组学分析了脂肪体Stat92e缺失和未缺失的Yki果蝇的相关代谢物水平,并将其与对照果蝇进行比较,结果发现丙氨酸、苯丙氨酸、亮氨酸和异亮氨酸水平的降低,这些水平通过耗尽Stat92e完全或部分恢复。为了进一步研究这些氨基酸的系统性缺乏是否由于脂肪体中糖异生的增加,接着对腹部样本进行代谢组学分析,发现包括丙氨酸、苯丙氨酸、亮氨酸、异亮氨酸和蛋氨酸在内的几种氨基酸含量减少,但可以通过抑制脂肪体中的JAK-STAT信号来挽救。这些结果表明,这些氨基酸被用于Yki果蝇的三羧酸(TCA)循环。在这些氨基酸中,异亮氨酸和蛋氨酸可以转化为琥珀酰辅酶A,可能导致α-酮戊二酸含量增加,同时ScsβA(琥珀酰辅酶A处理所需的基因)水平上调。Yki果蝇脂肪体中hop/JAK和Stat92e的缺失都降低了ScsβA的表达水平,表明代谢物和ScsβA表达的变化与JAK-STAT信号的激活有关。总之,这些数据表明肝脏激活JAK-STAT信号会破坏脂肪体和其他宿主器官的局部氨基酸稳态。
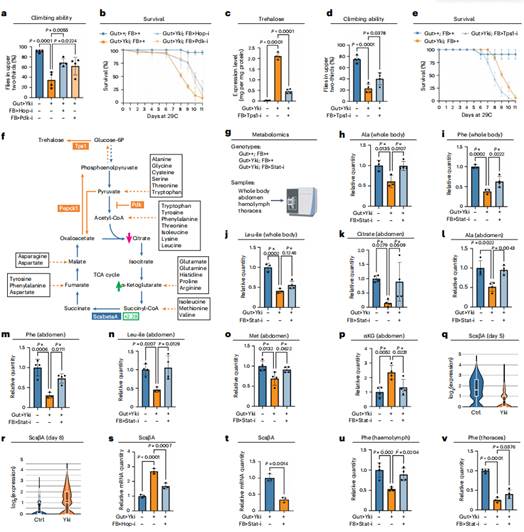
肝糖异生促进Yki果蝇的恶病质
为了在哺乳动物中验证这些发现,研究人员使用了一种完善的诱导性基因工程肺癌小鼠模型(KrasLSL-G12D/+;Lkb1flox/flox,简称KL小鼠)。研究人员假设癌症厌食症-恶病质综合征(CACS)KL小鼠中与恶病质相关表达的Pck1(果蝇Pepck1同源物)和Pdk3是通过相同的IL-6-JAK-STAT信号传导机制进行调节。用IL-6处理离体的小鼠原代肝细胞,只有较长时间的IL-6处理才能诱导Pck1和Pdk3的表达。值得注意的是,在注射产生IL-6的Lewis肺癌(LLC)细胞的小鼠(LLC+IL-6小鼠)中,肝脏中Pck1的表达似乎没有变化,而Pdk3是唯一上调的PDK同工酶。
为了将目前的结果扩展到人类,研究人员分析了基因型-组织表达(GTEx)项目中226个非病变肝脏样本的转录组学数据,发现PDK3的表达与IL-6-STAT3通路靶点以及转录因子基因STAT3呈正相关,这表明IL-6-JAK-STAT信号调节人类PDK3的表达。为了探索肝脏PDK3上调的不良影响,接着分析癌症基因组图谱(TCGA)项目中372名参与者的肝细胞癌队列的转录组数据。与小鼠模型结果一致,PDK3是唯一表达升高与生存率降低显著相关的同工酶,这表明肝脏PDK3表达增加导致预后不良,这可能是其在驱动代谢改变中发挥作用的结果。总之,研究结果支持IL-6-JAK-STAT信号促进肝脏PDK3表达的保守机制,从而导致与癌症相关的死亡率。

肿瘤诱导的JAK-STAT信号的保守性恶病质作用
总之,该研究通过利用多模型方法对癌症恶病质的发病机制提供了见解,揭示了Upd3-JAK-STAT或IL-6-JAK-STAT信号在癌症相关代谢紊乱中的保守致病作用,强调了靶向肝脏糖异生或PDK3在IL-6驱动的癌症恶病质中的潜在治疗策略。


